

2University Hospital “St. George”- Plovdiv, Bulgaria
3Sofia University “St. Kliment Ohridski”, Faculty of Medicine, Department of Biology, Medical Genetics and Microbiology, Sofia, Bulgaria
4Gynecology and Assisted Reproduction Hospital “Dr. Malinov”; Sofia, Bulgaria
5Medical University – Sofia, Department of Medical Genetics, Bulgaria
Abstract
Interstitial microdeletions of the distal 2p are very rare. A small number of cases have been reported in the literature, involving regions 2p23-p25, 2p23-p24 and 2p24-p25. The most common symptoms involve: intrauterine growth retardation, developmental delay, mental retardation, microcephaly, craniofacial anomalies, musculoskeletal abnormalities, congenital heart defect and hearing impairment. Herein we report on a Caucasian girl, born after in vitro fertilization with discrete facial dysmorphism, growth failure, borderline neurodevelopment and congenital heart defect. A de novo pericentric inversion of chromosome 2 was identified by routine karyotyping. An interstitial microdeletion of 2p24.3p25.1 was found by array karyotyping and following FISH analysis revealed that the deletion affects the inverted chromosome 2. This case illustrates the utility of high resolution methods to identify submicroscopic quantitative changes in structurally rearranged chromosomes. The precise determination of the genetic content of small quantitative changes in the genome provides important information for genetic counseling, enabling to predict the course of disease and the planning of adequate therapy and prophylaxis in affected families.
Citation: Ivanov HY, Stoyanova VK,. Vazharova R, Linev A, Ivanov I, Ivanov S et al. A de novo microdeletion 2p24.3-25.1 identified in a girl with global development delay. Biodiscovery 2015; 18: 1; DOI: 10.7750/BioDiscovery.2015.18.1
Copyright: © 2015 Ivanov et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, provided the original authors and source are credited.
Received: August 26, 2015; Accepted: October 08, 2015; Available online /Published: October 10, 2015
Keywords: microdeletion 2p24.3-25.1, global development delay
*Corresponding Author: Vili K. Stoyanova, email: vi1sto@abv.bg
Conflict of Interests: No potential conflict of interest was disclosed by any of the authors.
#These authors contributed equally to this work.
Introduction
Constitutional interstitial deletions of the distal short arm of chromosome 2 are very rare. A small number of cases have been described in the medical literature, involving regions 2p23-p25, 2p23-p24 and 2p24-p25 [1]. The most common symptoms involve: intrauterine growth retardation; developmental delay; mental retardation; microcephaly, craniofacial anomalies; facial dysmorphism (hypotelorism/hypertelorism, epicantal folds, narrow, short palpebral fissures, large, low-set ears), musculoskeletal abnormalities; congenital heart defect, seizures and hearing impairment.
We report on a girl with discrete facial dysmorphism, global development delay and congenital heart defect. A de novo pericentric inversion of chromosome 2 was found by routine karyotyping and an interstitial microdeletion 2p24.3-p25.1 was identified by array karyotyping.
Clinical Report
The proband is a Caucasian girl who came to our genetic clinic because of dysmorphism, growth failure and suspected neurodevelopmental delay. She was born at 37th gestational week of a twin pregnancy after in vitro fertilization with birth weight 1550 g., body length of 37 cm and head circumference of 29 cm. Her twin sister weighed 2200 g at birth and develops normally thus far. The pregnancy was complicated by bleeding in 6th m.l. Intrauterine growth retardation was noted at 35 g.w. Congenital heart defect (three apical ventricular septal defects) was found shortly after birth. Initial physical examination demonstrated axial musclе hypotonia.At the time of examination the proband is 1 year and 5 months old. Her weight is 6 kg (-3.23 SD), height is 64 cm (-3.9SD) and her head circumference is 39 cm (-4.38 SD).
Dysmorphic features include: hypertelorism, epicanthus, hirsutism of the forehead, prominent metopic suture, depressed nasal bridge, down slanting mouth corners, low set large and simple ears, high arched palate, misaligned dentition, carious teeth, short neck and low hairline at the neck. Her fingers are short with flexion contractures, clinodactyly of the fifth fingers and proximally placed thumbs and toes (Fig. 1). Neurological examination revealed a musclе hyperotonia of lower limbs. Her neurodevelopment was borderline:: able from prone to supine at 6 months of age; pronounce syllables at 1 year of age; first word at 13 months of age; able to walk with support at 17months.. Developmental delay – DQ – 71% by Manova-Tomova. Palm X-ray for bone age scan at chronological age 17 months revealed bone age of 1 month age. Abdominal echography showed normal liver, spleen and kidneys. Echocardiography detected ventricular septal defect. The thyroid function is normal.

Figure 1. Phenotype of the proband. Note the prominent metopic suture, hypertelorism, epicanthus, hirsutism of the forehead, down slanting mouth corners, low set large and simple ears the short fingers with flexion contractures, clinodactyly of the fifth finger and proximally placed thumb.
Cytogenetic and Molecular Analysis
Cytogenetic analysis of cultured blood lymphocytes was performed using standard protocols. In all metaphases a female karyotype with a large pericentric inversion of chromosome 2 was found – 46XX,inv(2)(p24q14.3)(Fig. 2). The inversion is apparently balanced at 400 GTG bands resolution. The karyotypes of the parents appeared normal at 400 bands resolution. The karyotype of the proband’s healthy twin-sister has not been analyzed. Because of the “chromosomal phenotype” of the patient the presence of a submicroscopic chromosomal imbalance was suspected.

Figure 2. (A) Partial GTG banded karyotype of the patient showing pericentric inversion of chromosome 2, the rearranged chromosome is on the right. (B) Ideogram of the inverted chromosome 2.
Molecular karyotyping
To investigate the DNA of the patient for submicroscopic deletions and duplications, the Illumina Human CytoSNP-12 microarray, iScan system and the KaryoStudio 1.4.3.0 software were used according to the manufacturer’s instructions. The analysis revealed a heterozygous interstitial microdeletion encompassing 4.46 Mb in the short arm of chromosome 2 with breakpoints in bands 2p24.3 and 2p25.1 – arr[hg19] 2p25.1p24.3(8,822,787-13,462,596)x1 (Fig. 3).

Figure 3. Deleted region of the patient is presented compared to CNVs reported in the DECIPHER database. Only 4 cases show deletions overlapping the region observed in the present case. Deletions are visualized as red bars and duplications as blue bars, corresponding GTG bands are shown on the top.
In order to validate the presence of the microdeletion and to determine if it affects the inverted chromosome 2 we performed FISH analysis using directly labelled custom probes (RP11-327F6 for 2p25.1 labelled with Cy3.5 (SO) and RP11-425I16 for 2q11.2 labelled with FITC (SG)). All interphase nuclei demonstrated two green signals but only one orange signal. In all analyzed metaphases it was obvious that the deletion affects the inverted chromosome 2 (Fig. 4).
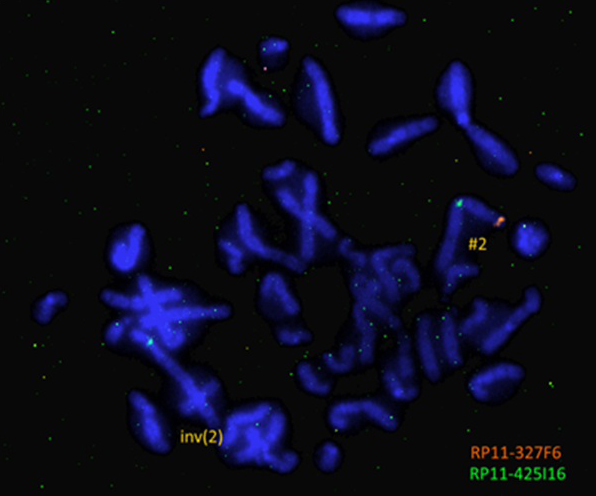
Figure 4. FISH analysis of the patient showing deletion of 2p25.1 (one orange signal for the probe RP11-327F6, and two green signals for the probe RP11-425I16) and location of the probe RP11-425I16 (2q11.2) in the ”short” arm of the derivative chromosome 2.
Discussion
Interstitial deletions of the distal 2p are very rare as constitutional abnormalities of the genome. There have been several reports of deletions encompassing the distal part of the short arm of chromosome 2 [1-7] but the number of patients with overlapping deletions is relatively small and a specific recognizable phenotype remains to be elucidated. A comparison of clinical findings in the present case and previously reported patients with overlapping interstitial deletions of the distal 2p is presented in Table 1. Mental retardation / developmental delay have been reported in all patents but with a variable degree of severity ranging from mild to severe. Intrauterine growth retardation / failure to thrive, anomalies of hands and feet, microcephaly / craniofacial anomalies and congenital heart defects have been observed in most patients sharing deletion of 2p24p25. The phenotypic heterogeneity may be the result of diagnostic difficulties in the identification and exact localization of 2p deleted segments in the prearray era [3, 4, 7]. Number of patients studied by molecular karyotyping is limited and present case contributes to a better understanding of genotype-phenotype correlation in carriers of subtle deletions of distal 2p.
Fifty two genes have been mapped to the region deleted in the patient. Among these genes 3 have been already implicated in monogenic human diseases: ADAM17 (autosomal recessive form – Inflammatory skin and bowel disease, neonatal, OMIM# 614328), KLF11 (autosomal dominant form of Maturity-onset diabetes of the young, type VII, OMIM# 610508), and LPIN1 (autosomal recessive form of acute recurrent Myoglobinuria, OMIM# 268200).
The deletion probably led to disruption of the gene KIDINS220 (kinase D-interacting substrate, 220kDa) and to a heterozygous loss of at least 1st to 22th exon of the gene. So far there are not any detected mutations of this gene associated with a particular disease in humans, but research on mouse models provides an association between change in gene dose and abnormal development of the heart and nervous system [8].
Some of the genes mapped to the region are predicted to be dosage-sensitive. ITGB1BP1 encodes Integrin beta-1-binding protein 1, which has been found to be involved in cell proliferation, differentiation, adhesion and migration in the context of the mineralization and development of bones and blood vessels, stimulating cell proliferation in a fibronectin-dependent manner. The loss of one copy of the gene could affect the development of bones and blood vessels. YWHAQ encodes the protein 14-3-3-THETA and is expressed in all tissues, with highest expression in the CNS. So far there are not any detected mutations in the gene associated with a particular disease, but a change in its expression is observed in patients with amyotrophic lateral sclerosis [9]. RRM2 encodes the small subunit (R2) of the enzyme ribonucleotide reductase which mediates the rate-limiting step in the synthesis of deoxyribonucleotides. Haploinsufficiency of the gene could be associated with retention in physical development observed in the patient. ROCK2 encodes RHO-associated coiled-coil-containing protein kinase 2. According to studies in animal models it has been found that loss of the gene leads to ROCK2 placental dysfunction and may cause delay in intrauterine fetal growth and even fetal death [10]. Recently, the gene has been identified as one of the candidate genes for heterotaxy in humans [11]. Another gene in the region – PDIA6, encodes the enzyme disulfide isomerase, family A, member 6, which is involved in the establishment of left-right symmetry during embryonic development and is important for the formation of the internal organs in the midline (heart, liver, pancreas) [12].
Conclusion
In conclusion we can say that this case illustrates the utility of high resolution methods in order to identify subtle quantitative changes in genome of patients with microscopically balanced chromosomal rearrangements. The precise determination of the genetic content of submicroscopic imbalances of the genome provides important information for genetic counseling, enabling to predict the course of disease and to plan adequate therapy and prophylaxis in affected families. As molecular karyotyping results become more frequently reported, a genotype–phenotype correlation based on contents and specific location may give clinicians further guidance for the diagnosis and prognosis of similar chromosome anomalies.
References
- Su PH, Chen JY, Tsao TF, Chen SJ. De novo interstitial deletion of chromosome 2 (p23p24). Pediatr Neonatol. 2011 Feb; 52(1): 46-50.
Reference Link - Neidich J, Zackai E, Aronson M, Emanuel BS. Deletion of 2p:a cytogenetic and clinical update. Am J Med Genet 1987 Jul; 27(3): 707-10.
Reference Link - Emanuel BS, Zackai EH, Van Dyke DC, Swallow DM, Allen FH, Mellman WJ. Deletion mapping: further evidence for the location of acid phosphatase (ACP1) within 2p23. Am J Med Genet 1979; 4: 167e72.
- Neidich J, Zackai E, Aronson M, Emanuel BS.Deletion of 2p: a cytogenetic and clinical update. Am J Med Genet 1987; 27: 707e10.
- Francis GL, Flannery DB, Byrd JR, Fisher ST. An apparent de novo terminal deletion of chromosome 2 (pter/p24:). J Med Genet 1990; 27: 137e8.
- Saal HM, King LJ, Zimmerman D, Johnson RC, Carr AG, Samango-Sprouse CA et al. Loss of the N-myc oncogene in a patient with a small interstitial deletion of the short arm of chromosome 2. Am J Med Genet 1996; 66: 373e7.
- Penchaszadeh VB, Dowling PK, Davis JG, et al. Interstitial deletion of chromosome 2 (p23p25). Am J Med Genet 1987; 27: 701e6.
- F Cesca, A Yabe, B Spencer-Dene, A Arrigoni, M Al-Qatari, D Henderson et al Kidins220 / ARMS is an essential modulator of cardiovascular and nervous system development, Cell Death Dis. 2011 Nov; 2(11): e226.).
Reference Link - Malaspina, A., Kaushik, N., de Belleroche, J. A 14-3-3 mRNA is up-regulated in amyotrophic lateral sclerosis spinal cord. J. Neurochem. 2000; 75: 2511-2520.
Reference Link - Thumkeo, D., Keel, J., Ishizaki, T., Hirose, M., Nonomura, K., Oshima, H. et al., Targeted disruption of the mouse Rho-associated kinase 2 gene results in intrauterine growth retardation and fetal death. Molec. Cell. Biol. 2003; 23: 5043-5055.
Reference Link - Fakhro, KA, Choi, M., Ware, SM, Belmont, JW, Towbin, JA, Lifton, RP et al.. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc. Nat. Acad. Sci. U S A 2011; 108: 2915-2920.
Reference Link - Hoshijima, K., Metherall, JE, Grunwald, DJ A protein disulfide isomerase expressed in the embryonic midline is required for left / right asymmetries. Genes Dev. 2002; 16: 2518-2529.
Reference Link

